

Lessons from Humira
Lessons from Humira
What I learned from diving into the preclinical development and clinical trial timeline for one of the biggest blockbuster drugs.

I. Introduction
I've written some abstract position pieces recently, especially around "how biotech can be better". While I enjoy exploring big ideas, it also ignites my fear that I'm turning into one of those thinkbois who loses touch with reality. To counteract this tendency, I decided it was time to do something on the opposite end of the spectrum: dig into a very specific, important case and see what concrete takeaways emerge.
Enter Humira (adalimumab), the first fully human monoclonal antibody therapy and one of the best-selling drugs of all time. Approved in 2002 for rheumatoid arthritis, Humira and other branded versions of the same molecule have since become a cornerstone treatment for various autoimmune diseases. It's also the drug that turned Abbott (now AbbVie) into a top 5 biopharma company by market cap. Beyond its commercial success, Humira interests me as a concrete example of what developing a novel, breakthrough therapeutic in a new modality looks like.
Let's dive in.
II. Humira Overview
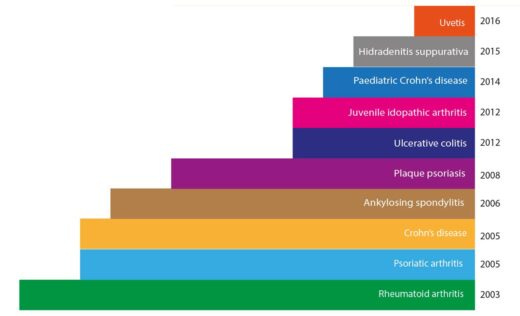
As the first fully human monoclonal antibody approved for therapeutic use, Humira was a big leap forward in our ability to harness the immune system to fight disease. Initially approved by the FDA in 2002 for the treatment of rheumatoid arthritis, Humira's since had its approval extended to a wide range of autoimmune conditions, including Crohn's disease, ulcerative colitis, psoriasis, and ankylosing spondylitis, among others. This broad applicability across multiple diseases is a key factor in its extreme commercial success.
Humira has consistently ranked as one of the world's best-selling drugs, with annual sales peaking at nearly $21 billion in 2022 (Humira revenue over time). This financial windfall transformed its parent company, Abbott Laboratories (which later spun off its pharmaceutical business as AbbVie), propelling it into the ranks of top global pharmaceutical companies.
But Humira's not just a financial success. For millions of patients, it has offered relief from debilitating autoimmune conditions, improving quality of life in ways that are hard to quantify. Its success also paved the way for a wave of other monoclonal antibody therapies, cementing biologics as a major class of drugs in modern medicine.
III. How Humira Works
At its core, Humira works by targeting and neutralizing a protein called tumor necrosis factor alpha (TNF-α). TNF-α’s name is an artifact of its discovery rather than a true description of its full scope. Despite its name suggesting a role in tumor biology, TNF-α is actually a key player in the body's inflammatory response. To understand why this matters, we need to take a brief detour into the world of cytokines and autoimmune diseases.
Cytokines are small proteins that act as messengers in our immune system. They can trigger inflammation, activate immune cells, and coordinate complex immune responses. Under normal circumstances, this is a good thing – inflammation is a crucial part of our body's defense against pathogens and tissue damage.
However, in autoimmune diseases like rheumatoid arthritis, the immune system goes haywire. It starts attacking the body's own tissues, leading to chronic inflammation. In these conditions, TNF-α doesn't just participate in the inflammatory response – it often plays a starring role, kicking off a cascade of inflammatory signals that perpetuate the disease.
This is where Humira comes in. As a monoclonal antibody, Humira is a protein designed to bind specifically to TNF-α. When Humira binds to TNF-α, it prevents this inflammatory cytokine from interacting with its receptors on cells. In essence, Humira acts like a sponge, soaking up excess TNF-α and reducing the overall inflammatory signal in the body.
By dampening this key inflammatory pathway, Humira can significantly reduce symptoms in a variety of autoimmune diseases. It's not a cure – patients typically need ongoing treatment to maintain its effects – but for many, it can dramatically improve quality of life and slow disease progression.
Understanding this mechanism is crucial to appreciating the scientific journey behind Humira's development. The road to creating this drug involved not just developing the antibody itself, but also unraveling the complex role of TNF-α in autoimmune diseases. As we'll see in the next section, this process drew on advances in multiple fields of biology and biotechnology.
IV. Key Enabling Developments
Humira's development was the result of several converging lines of research and technological advances. Three were especially important:
A. Understanding Cytokine Cascades and Rheumatoid Arthritis
The story of Humira begins with basic research into the mechanisms of inflammation and autoimmune disease. In the 1980s, researchers began to unravel the complex network of signaling molecules involved in inflammation, particularly in rheumatoid arthritis (RA).
A pivotal moment came in 1989 when a team led by Marc Feldmann and Ravinder Maini at the Kennedy Institute of Rheumatology in London made a crucial discovery. Their research, which involved analyzing mRNA expression in RA joint cells, revealed persistent expression of interleukin-2 (IL-2) and its receptor, as well as interferon gamma (IFN-γ). This suggested that prolonged T cell activation and cytokine production played a key role in RA pathogenesis.
Building on this work, Feldmann and Maini's team later identified TNF-α as a central player in the inflammatory cascade. In a simplified view of RA, TNF-α triggers inflammation in the joints, setting off a chain reaction. This immune response initiates a cytokine cascade that ultimately leads to the breakdown of collagen and other structural components in the joints.
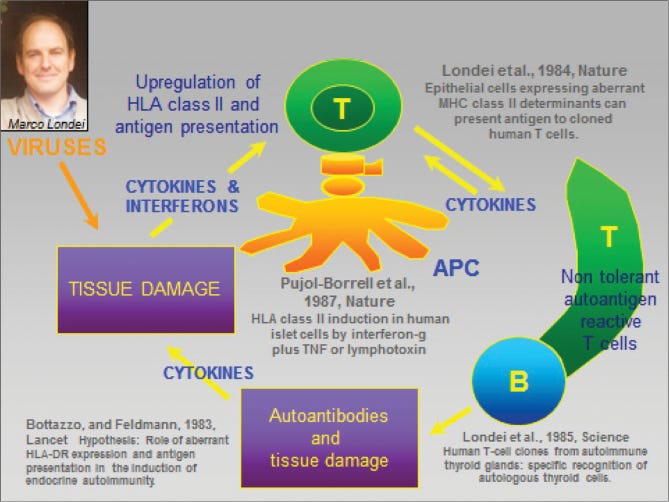
This insight was a big deal because it provided an important leverage point for intervention. It suggested that targeting a single molecule – TNF-α – could potentially treat a complex disease like RA by interrupting this destructive cascade. This provided a clear rationale for developing anti-TNF-α therapies.
B. Monoclonal Antibody Technology
The idea of using antibodies as therapeutic agents had been around since the 1970s (early paper), but it took decades to turn this concept into a practical reality. The key breakthrough came in 1975 when Georges Köhler and César Milstein developed a technique for producing monoclonal antibodies – identical antibodies that all target the same specific molecule.
However, early therapeutic antibodies were derived from mice, which led to problems when used in humans. The human immune system would recognize these mouse antibodies as foreign and attack them, limiting their effectiveness and potentially causing adverse reactions.
C. Phage Display and Antibody Humanization
The final piece of the puzzle was developing a way to create fully human antibodies. This is where phage display, a technique developed by George Smith in 1985 and further refined by Greg Winter and his colleagues in the 1990s, came into play.
Phage display allows researchers to create vast libraries of antibodies and rapidly screen them to find those that bind most effectively to a target molecule. To understand why it was an important development, it’s helpful to understand the problem phage display solves. Measuring the function (eg, binding) of antibodies or any protein is hard to do at scale because we don’t have a way to sequence proteins. This means we don’t have a good way to directly take a set of proteins in an assay and figure out how many of the different variants we started with and ended up with, the basic principle behind many biological enrichment assays. As a result, without something like phage display, we’d instead remain stuck with measurement methods where we keep variants of proteins in separate wells/dishes/pools, limiting us to 100s of simultaneous measurements.
Phage display lets us scale up well beyond this by taking advantage of a Trick So Simple You’ll Wish You Thought Of It. Phage display takes advantage of two properties of phages:
They package their own genomes.
They will display protein fragments added to their genome on their surface.
Putting these together enables the multiplexed measurement of antibody binding. It works as follows. We start with a set of antibody fragment variants for which we wish to measure binding to a specific target protein (antigen). To measure binding using phage display, we first use relatively basic molecular cloning techniques to clone these antibody variants’ DNA into (a specific position within) plasmids containing phage genomes. This gives us plasmids containing phage DNA and bacterial fragments embedded within the former.
Next, we transfect bacteria with these plasmids, resulting in lots of phage particles bursting from these bacteria. Here’s where the two previously mentioned properties come in. If everything goes right, these phage particles will now have antibody fragments displayed on their surface and have the DNA encoding said fragments packaged inside as part of their self-packaged DNA.
To measure binding, we take our phage particles and flow them over a bunch of proteins fixed onto a surface. Those that bind well will stick even with washing, whereas the weak binders will fall off. From here, we can determine which variants are the ones that stick by taking advantage of self-packaging. We lyse and sequence the variants that are stuck and then identify the antibody fragment DNA within the sequenced genomes to determine which variants are bound.
That’s a simple description, so I’m unsurprisingly leaving out a lot of detail as well as variants of the protocol, but that gives you enough to understand the key idea. The basic idea is that the large number of phages, the ease of measuring antigen binding, and the ability to recover the DNA strand associated with stronger binding, combine to give us a multiplex way to measure antibody binding.
For developing Humira, phage display was used as part of a humanization protocol to convert known TNF-α binding antibodies from mice into human versions. I’ve described this briefly below, but if you want to read more, you can check out this review of humanization by guided selection.
The process started with mouse antibodies that effectively bound to TNF-α. They could get these mouse antibodies because they could immunize mice with TNF-α and then harvest their antibodies – a process that would be unethical in humans.
These mouse antibodies then underwent a process called "guided selection" to humanize them. This step is crucial because human immune systems would recognize mouse antibodies as foreign and attack them, limiting their effectiveness and potentially causing adverse reactions.
In guided selection, components of genes for the mouse antibodies' binding regions are inserted into phage display libraries of other components human antibody genes. For example, one sub-library might take light chain fragments from the mouse antibodies and mix them with human heavy chain fragments. These hybrid antibodies are then subjected to rounds of selection, each time choosing the variants that bind best to TNF-α.
Over several rounds, the mouse components are gradually replaced with human sequences that maintain or improve binding to TNF-α and recombined. The result is a fully human antibody that retains the specific binding properties needed to target TNF-α effectively.
Winter and his team at Cambridge Antibody Technology (CAT) used this technique to develop the antibody that would become Humira. This approach solved the immunogenicity problem of earlier antibody therapies while maintaining their therapeutic potential.
These three developments – understanding the role of TNF-α in RA, the ability to produce monoclonal antibodies, and the technique to create fully human antibodies – converged to make Humira possible. The next section puts these different developments on a linear timeline to show how long things took and how they came together.
V. Timeline of Critical Milestones
The development of Humira spans several decades, from foundational research to clinical trials and approval. In order to force myself to reason from the concrete details, I found it helpful to construct a detailed timeline of its development, broken down into a few sections. I’ve also included an even more detailed timeline in the appendix for people who want to go deeper.
1974-1989: Foundational Research and Technology Development
1975: Köhler and Milstein develop the hybridoma technique for producing monoclonal antibodies.
1984: The TNF gene is characterized and cloned, enabling further study of its role in inflammation.
1985: George Smith demonstrates phage display, laying groundwork for future antibody development techniques.
1989: Feldmann and Maini publish their work on cytokine expression in rheumatoid arthritis synovial tissue.
1989-1995: Translation to Therapeutic Application
1991: Greg Winter and colleagues demonstrate the production of human antibodies in transgenic mice.
1993: BASF Pharma commissions Cambridge Antibody Technology (CAT) to produce fully human antibodies to TNF-α.
1994: Winter and colleagues publish on making human antibodies by phage display technology.
1995: Tristan Vaughan at CAT isolates D2E7, the antibody that would become adalimumab, using phage display.
1995-2002: Preclinical and Clinical Development
1997-1998: Phase I clinical trials of D2E7 begin.
1998-2001: Phase II and III clinical trials are conducted, demonstrating efficacy and safety in rheumatoid arthritis patients.
2002-2003: Approval and Launch
December 31, 2002: The FDA approves Humira for the treatment of rheumatoid arthritis.
January 2003: Humira is launched in the US market.
VI. Analysis: What Took So Long?
Looking at Humira's development timeline, the question I was most interested in was: why did it take nearly three decades from the initial breakthroughs in monoclonal antibody technology to Humira's approval? To answer this, let's break down the major phases of Humira's development and examine what contributed to the lengthy process.
A. Discovery: Understanding TNF-α and Autoimmunity
Contrary to my initial expectations, the most time-consuming aspect of Humira's development wasn't clinical trials – it was the discovery process. This phase took around 15 years, depending on where you start and end the clock, and required a series of sequential insights, each building on the last.
Getting to TNF-α as a reasonably good hypothesis for a target required:
Understanding the role of cytokines in inflammation
Identifying TNF-α as a key player in rheumatoid arthritis
Demonstrating that blocking TNF-α could suppress the inflammatory cascade
Each of these steps required careful experimentation and analysis. And these insights weren't obvious at the outset – they emerged gradually as researchers pieced together the complex puzzle of autoimmune disease.
The serial nature of these discoveries means that parallelization wasn't really always possible. You can't start developing a TNF-α inhibitor until you know that TNF-α is a good target, and you can't know that until you understand its role in the inflammatory cascade.
B. Tech Dev: From Mouse to Human Antibodies
While the scientific insights were accumulating, the technology to create humanized therapeutic antibodies was also evolving. Getting from mouse antibodies to fully human antibodies like Humira involved several steps:
Developing hybridoma technology for producing monoclonal antibodies
Creating chimeric antibodies (part mouse, part human)
Figuring out how to create humanized antibodies
Finally, creating fully human antibodies through phage display
Similar to the discovery process, these developments built on each other. For example, phage display's potential was partially realized through seeing the limitations of just developing antibodies in mice.
C. Clinical Development: The Final Stretch
Once the target was identified and the technology to create human antibodies was available, Humira still had to go through the standard drug development process: preclinical animal model experiments followed by three, partly parallelizable phases of trials.
While this process took about 7 years for Humira – not unusually long for drug development – it comes at the end of a much longer period of foundational research and technology development.
Summing up, while they took a while (~7 years), trials weren't the most time-consuming part of Humira's development; it was the foundational scientific work.
The Cost of Unnecessary Slowdowns
On the flip side, Humira's development hammers home the potential impact of unnecessary scientific slowdowns. Given the serial nature of insights, unnecessary delays in the research process can have outsized impacts. For instance, if peer review or publication delays did add even a year to each major step in understanding the TNF-α pathway, it could have significantly extended Humira's overall development timeline. This suggests that finding ways to accelerate knowledge sharing in the scientific community could have substantial benefits.
VII. Key Takeaways and Broader Implications
Digging into Humira's development history has left me with several hypotheses that may extend beyond this specific case. That said, I am worried about over-generalizing, so these should be taken as speculative and provisional rather than conclusive.
A. Trials aren’t (always) the primary bottleneck in therapeutics development
Before writing this, I had the, in hindsight not sufficiently pressure tested, impression that trials were typically the most time-consuming step of drug development. As mentioned already, that wasn’t the case for Humira. The fundamental research and tech development took roughly twice as long as the trials.
This left me wondering how off base my previous impression was. In particular, it's hard for me to know how well this generalizes given Humira was a leader in a new therapeutic modality and disease area. Fortunately, I consulted a friend who works in the trials space, and they provided some relevant context:
Low hanging fruit for speeding up trials are likely indications where incremental improvements are possible, relatively unexplored problem spaces that have high patient burden, and first-in-class (“breakthrough” as categorized by the FDA) treatments for specialized populations.
On the other hand, treatments that are more general such as Humira as well as Ozempic (GLP-1 agonists) often have an easier time recruiting for trials while also having longer foundational scientific incubation times.
Based on this, my updated view is that trials are still a major bottleneck, but not in every single case. In particular, for high impact therapeutics based on scientific breakthroughs, novel technologies, and/or modalities, all the scientific and technological fundamentals reaching maturity will take longer than the subsequent trials.
B. Rethinking Progress Metrics in Biotech: Is Eroom's Law a Red Herring?
Humira's story has me reconsidering the value of ideas like "Eroom's Law" for quantifying progress in biotech. While metrics like the number of new drugs approved per year (as captured in Eroom's Law) matter, they might not fully capture the impact of truly breakthrough therapies. Humira, for instance, paved the way for an entire class of biologics that have transformed autoimmune disease treatment, but depending on how you count, it would only register as 1 new drug in two decades.
This suggests we should be cautious about drawing conclusions from high-level statistics without digging into the details of individual cases. As we see with Humira, a single high-impact therapy can potentially be more valuable than multiple incremental improvements. But a too-strong focus on optimizing something like "reversing Eroom's" could steer us towards the latter.
I'm not saying we should ditch metrics altogether. Measures like new drug approvals are important, but we should balance them with metrics that capture the impact of truly breakthrough therapies. This might involve tracking quality of life improvements for patients with previously intractable conditions, or measuring scientific and technological spillover effects from major innovations.
Taking inspiration from tech, the ultimate form of a progress dashboard in biotech may look more like the Weekly Business Review than a single north star metric. Briefly, although I really recommend reading the article (I know it’s long), this would mean monitoring many input metrics (number of drugs approved, type of drugs approved, trial success rates, spread across modalities, costs of research, etc.) that have been tested and refined to correspond to output metrics (e.g. number of diseases cured) rather than just focusing on a single number.
C. The Value of Comprehensive Historical Records and Oral Histories
In writing this piece, I relied heavily on publicly available papers and other historical documents to reconstruct Humira's timeline. But I often found myself wishing I could supplement with stories from the key people involved to get more nuance. For example, earlier I hypothesized that unnecessary slowdowns could have significantly impacted Humira's development time. But that's just a hypothesis - I don't actually know how many unnecessary slowdowns were involved. It's equally possible that all the key researchers were in constant communication, making peer review timelines irrelevant, or that there was a 2-year stall that could have been fixed with the right 5-minute conversation.
This makes me think producing more oral histories for important scientific developments could be a low-hanging fruit win for meta-science. It costs relatively little but can fill in those gaps in our understanding of history that can significantly affect the conclusions we draw. Paraphrasing John Salvatier, history is surprisingly detailed!
D. Long-Term Investment in High-Impact Therapeutic Innovations
Having recently learned about the history of GLP-1 agonists, it struck me that both they and monoclonal antibodies took multiple decades to develop, but then ended up being disproportionately high impact. (See Blake Byers on this topic here.) Similarly, if you start from the discovery of AAV, AAV gene therapy has a 50+ year development history, and even the most optimistic timeline would have it taking 2+ decades to go from early research to application. On the other hand, CRISPR-based therapies might represent a potential counterexample, having moved more quickly from discovery to the clinic. Still, this has me wondering if, especially now that we've picked therapeutic low-hanging fruit, we should expect that the most impactful therapeutics will involve long scientific discovery/tech development phases by default.
Beyond wanting to gather additional evidence for/against this, one obvious takeaway is that long-term investment horizons are important. A more counterintuitive takeaway might be that making science and tech dev cheaper is crucial because it reduces the investment needed to sustain this long-term focus.
Finally, the same friend mentioned above shared some helpful nuance here as well. While it’s true that Humira had such an extended development, Abbott Research (which eventually became AbbVie) was able to acquire Knoll Pharmaceuticals in 2000 and then develop Humira for so many other indications because they had a pipeline of other, lower risk, lower reward assets that made them a viable business. Similarly, Novo Nordisk’s support for much of the research that eventually led to Ozempic was of course funded by their existing insulin business. My takeaway from this is that one path to supporting such high risk, high reward, long payoff bets is to build a viable business around other less high variance bets and then use that to fund the riskier ones. Obviously this comes with organizational challenges, but it does seem to have worked in at least a few cases.
If anyone has other thoughts/evidence related to this point, it's one I'm especially interested in hearing about.
Conclusion
Digging deep into Humira's history has been a valuable exercise, although, as cliché as it sounds, it's left me with more questions than answers. I've still come away with a few provisional insights that I think are worth highlighting though:
Trials aren't always the biggest time-sink. For Humira, the real time drain was the basic science and tech development. First-in-class, scientifically novel, general therapeutics in particular may often require long incubation times and therefore be relatively less bottlenecked by trials.
Our metrics for biotech progress might need rethinking. Something like Humira, which paved the way for a whole new class of drugs, only counts as one approval in the usual stats. Perhaps we need to reconsider how we measure impact in this field.
There might be something to the idea that truly groundbreaking drugs take longer to develop. Humira, GLP-1 agonists, AAV gene therapy - they all took decades. Is this a pattern, or just cherry-picking? I'm not sure yet, but it's got me thinking.
In terms of follow-up, I'm particularly interested in digging into whether Humira represents an exception that proves the rule with respect to trials being less of a bottleneck, and this nascent idea about impactful therapeutics having longer development times. Both feel like ideas that would be quite "big if true" for my views on the biotech industry and levers to accelerate it.
As always, if you have thoughts, questions, or disagreements, don't hesitate to comment or reach out to me. This stuff is complex, and I'm always open to hearing different perspectives or being told I'm off base.
Acknowledgments
Thanks to my friend who works on trials for their extremely helpful context and nuance, Willy Chertman for his helpful (as always) feedback and edits, and Abhi “Owl Poster” Mahajan for high level feedback.
Appendix
Detailed Humira Timeline
1974: First fusion mouse-tumor cell line for manufacturing antibodies developed (paper),
1975: Initial discovery of TNF in endotoxin-dosed mouse serum as a tumor destroying factor (paper).
1978: Commercial distribution of fusion cell lines by MRC/Sera-Lab begins (source).
1984: Key hypothesis about the relationship between T-cell triggered ‘cytokine cascade’ and autoimmunity based on connection between viral infection and autoimmunity published (paper).
1984: TNF gene characterized and cloned (paper). Relevant for subsequent work to characterize the inflammatory response in rheumatoid arthritis and to identify TNF-ɑ as a good target.
1985: First successful demonstration of phage display (paper), the technology eventually used to develop adalimumab.
1986: Mouse antibody humanisation demonstrated (paper). Precursor to subsequent full humanisation of antibodies.
1988: Discovery that many pro-inflammatory cytokines including TNF-ɑ are produced in rheumatoid arthritis culture (paper)
1989: Target validation in rheumatoid arthritis culture with TNF-ɑ shown to inhibit cytokine cascade (paper)
1989: Cambridge Antibody Technology (CAT) founded to commercialize phage therapy, which was eventually used to discover adalimumab.
1991: Demonstration of human antibody production in transgenic mice (paper). Informed subsequent phage therapy humanization work.
1993: BASF pharma commissions CAT to produce fully human antibodies to TNF-α (source).
1994: Making human antibodies by phage display technology (Winter paper) and humanizing rodent antibodies (paper)
1995: Tristan Vaughan, a scientist at CAT, isolates a potential drug candidate known as D2E7 (which became adalimumab) using the phage display technology (source, paper describing the method).
1999: Preliminary positive results released from adalimumab (double blind) phase I trial (paper).
2001: Phase I and phase II trials conclude, confirming initial efficacy and dose toleration (phase I results, phase II results).
2002: Adalimumab (Humira) approved for treatment of Rheumatoid Arthritis in patients whose disease hasn’t responded to other treatments (FDA approval document).
2003: Humira launches.
2003-today: Subsequently approved for a wide range of autoimmune diseases
Nice! Something I'd add about the discovery process is that the safety of tnf-alpha inhibitors was not obvious and is still not well understood. Some big part of why they were hard to develop is that it's weird that blocking tnf-alpha can be effective in so many autoimmune diseases but doesn't seem to cause too much cancer or serious immunosuppression. I think a lot of effort was spent establishing that in animals. Also, this paper (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1766582/) suggests that they were really focused on a fully humanized antibody to have clearly better pk than the existing mabs, which sounds like it was pretty tricky. It reminds me of all the engineering that went into fixing semaglutide's pk.
Interesting comments regarding the importance (or lack thereof) of clinical trials as a rate-limiting step. However the cost of trials is rising inexorably (IQVIA), and the entire clinical process is arguably an oligopoly run by a small number of large CROs. This suggests a sector in need of real disruption. I’d also add that Abbvie is a pretty hard-nosed organisation that was determined to extract every $ of value. Consequently Humira enjoyed even greater success than expected. Thoughts on Eroom’s Law are certainly thought provoking although it’s clear far too early (in my opinion) to suggest that AI directed drug design has really moved the needle regarding POS.